STUDY DESIGN
TRIAL 4: A phase 3, open-label safety study in patients aged 2 to <6 years1-3,a
- Phase 3, 24-week, open-label study evaluating the safety and tolerability of TRIKAFTA2,3,a,b,c
- 75 patients received TRIKAFTA
- Dosage was based on weight:
- ≥10 to <14 kg (n=16): elexacaftor 80 mg/tezacaftor 40 mg/ivacaftor 60 mg qam and ivacaftor 59.5 mg qpm2,3
- ≥10 to <14 kg (n=16): elexacaftor 80 mg/tezacaftor 40 mg/ivacaftor 60 mg qam and ivacaftor 59.5 mg qpm2,3
- ≥14 kg (n=59): elexacaftor 100 mg/tezacaftor 50 mg/ivacaftor 75 mg qam and ivacaftor 75 mg qpm2,3
- ≥14 kg (n=59): elexacaftor 100 mg/tezacaftor 50 mg/ivacaftor 75 mg qam and ivacaftor 75 mg qpm2,3
- Dosage was based on weight:
aPatients on CFTR modulators were asked to washout for 4 weeks prior to Day 1.
bAll patients who completed the last treatment period visit and did not permanently discontinue the study drug were enrolled in the open-label Extension Study.
cTrial 4 was a 2-part study: Part A was a pharmacokinetic and safety study while Part B evaluated safety, pharmacokinetics, pharmacodynamics, and efficacy.
Select Inclusion Criteria1-3*
- Confirmed CF diagnosis
- Patients who had at least one F508del mutation or a mutation known to be responsive to TRIKAFTA were eligible for the study
- Stable CF as deemed by the investigator at screening
- Ages 2 to <6 years at Day 1 of the study
- Body weight ≥10 kg at screening
*Key exclusion criteria included clinically significant cirrhosis with or without portal hypertension, lung infection with organisms associated with a more rapid decline in pulmonary status (including, but not limited to, Burkholderia cenocepacia, Burkholderia dolosa, and Mycobacterium abscessus), and solid organ or hematologic transplantation.
Primary Endpoint2
- Safety and tolerability as determined by adverse events and clinical laboratory assessments
Select Secondary Endpoints2
- Absolute change from baseline through Week 24 in sweat chloride concentration
- Absolute change from baseline through Week 24 in LCI2.5
Select Additional Endpoints2
- Absolute change from baseline at Week 24 in BMI
- Absolute change from baseline at Week 24 in BMI-for-age z-score
Baseline Characteristics2
| TRIKAFTA (N=75) | |
|---|---|
| Sex, female, n (%) | 41 (54.7) |
| Mean age, years (SD) | 4.1 (1.1) |
| Mean sweat chloride, mmol/L (SD) | 100.7 (11.2) |
| Mean BMI, kg/m2 (SD) | 15.79 (1.06) |
| Mean BMI-for-age z-score (SD) | 0.09 (0.85) |
| Mean LCI2.5, units (SD) | 8.41 (1.48) |
Trial 4 Limitations and Disclosures2
- Trial 4 was an open-label study with no placebo-control arm designed to assess safety as the primary endpoint. Therefore, causality cannot be attributed to TRIKAFTA. (All patients in the study knew they were on active treatment, which may have introduced a bias related to that awareness.)2
- Parts of this study overlapped with the COVID-19 pandemic; therefore, some patients were unable to complete in-person evaluations at all time points. For patients who were unable to complete in-person visits, data were collected remotely and those patients remained in the study2
- Some results from Trial 4 are not included in the approved full Prescribing Information, and the FDA did not consider the study in the initial approval of TRIKAFTA
SUMMARY OF RESULTS
Trial outcomes for patients aged 2 to <6 years2
Primary Endpoint2
SAFETY AND TOLERABILITY OF TRIKAFTA
The safety profile of TRIKAFTA in patients aged 2 to <6 years is consistent with the safety profile established in Trial 11,2
Discontinuations and serious adverse events
- 2 patients (2.7%) experienced serious adverse events2
- One patient experienced abnormal behavior that led to study drug discontinuation. The patient had a history of behavioral and developmental issues, and developed hyperactivity, aggression, increased urinary urgency, and enuresis, which resolved after treatment discontinuation. The investigator assessed the events as possibly related to treatment
- One patient had a pulmonary exacerbation, which was considered not related to treatment and resolved without change in treatment
- There were no deaths in Trial 44
| TRIKAFTA n (%) (N=75) | |
|---|---|
| Patients with any TEAEs | 74 (98.7) |
| Cough | 46 (61.3) |
| Pyrexia | 26 (34.7) |
| Rhinorrhea | 25 (33.3) |
| Vomiting | 21 (28.0) |
| COVID-19 | 14 (18.7) |
| Nasal congestion | 13 (17.3) |
| Rash | 12 (16.0) |
| Upper respiratory tract infection | 11 (14.7) |
| Decreased appetite | 9 (12.0) |
| ALT increased | 8 (10.7) |
| Infective PEx of CF | 8 (10.7) |
Liver-related adverse events2
| TRIKAFTA n (%) (N=75) | ||
|---|---|---|
| Elevated ALT or aspartate transaminase (AST), n (%) | >3x ULN | 6 (8.0) |
| >5x ULN | 2 (2.7) | |
| >8x ULN | 1 (1.3) | |
| ALT or AST >3x ULN and total bilirubin >2x ULN | 0 | |
| Adverse events of elevated ALT and/or AST | 8 (10.7) | |
- 1 patient required treatment interruption during Trial 4 and later discontinued TRIKAFTA during the open label extension due to transaminase elevations1
Rash events2,3
- 15 patients (20.0%) experienced rash eventsd
- 12 patients had rash events that were assessed as unlikely/not related to study drug and/or were confounded by concurrent viral symptoms
- No patients discontinued due to rash events, although 2 patients had interruptions and both resumed study drug without recurrence of rash
- Rash events were more frequent among males (32.4%) than females (9.8%)
dRash events were determined to be mild or moderate in severity. Includes rash, rash erythematous, rash maculopapular, rash papular, and urticaria.
Select Secondary Endpoints

REDUCTION IN SWEAT CHLORIDE CONCENTRATION2
LS-mean absolute change from baseline through Week 24 (95% CI: -61.3, -54.6)
Mean (SD) at baseline: 100.7 mmol/L (11.2)
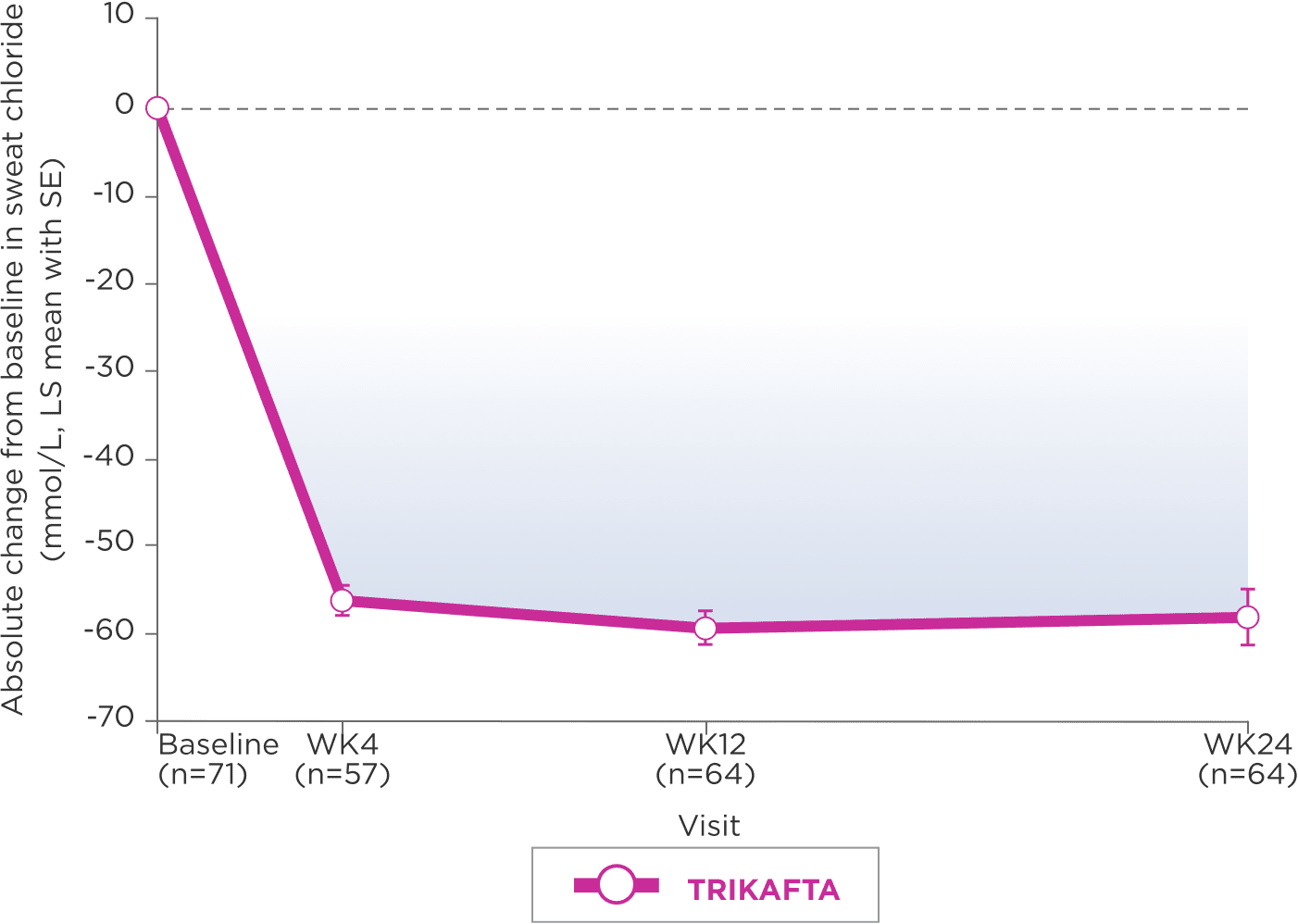
- Subgroup analysis of absolute change in sweat chloride by genotype saw LS mean (95% CI) reductions of 52.6 mmol/L (-56.9, -48.4) in patients with F/MF genotype (n=47) and -70.0 mmol/L (-75.4, -64.5) in patients with F/F genotype (n=22)2
eIn Trial 4, mean baseline sweat chloride (SD) was 100.7 mmol/L (11.2) for all patients receiving TRIKAFTA in this study.
85% of patients heterozygous for F508del and another specific mutation and 100% of patients homozygous for F508del mutation had sweat chloride concentrations of <60 mmol/L through Week 243
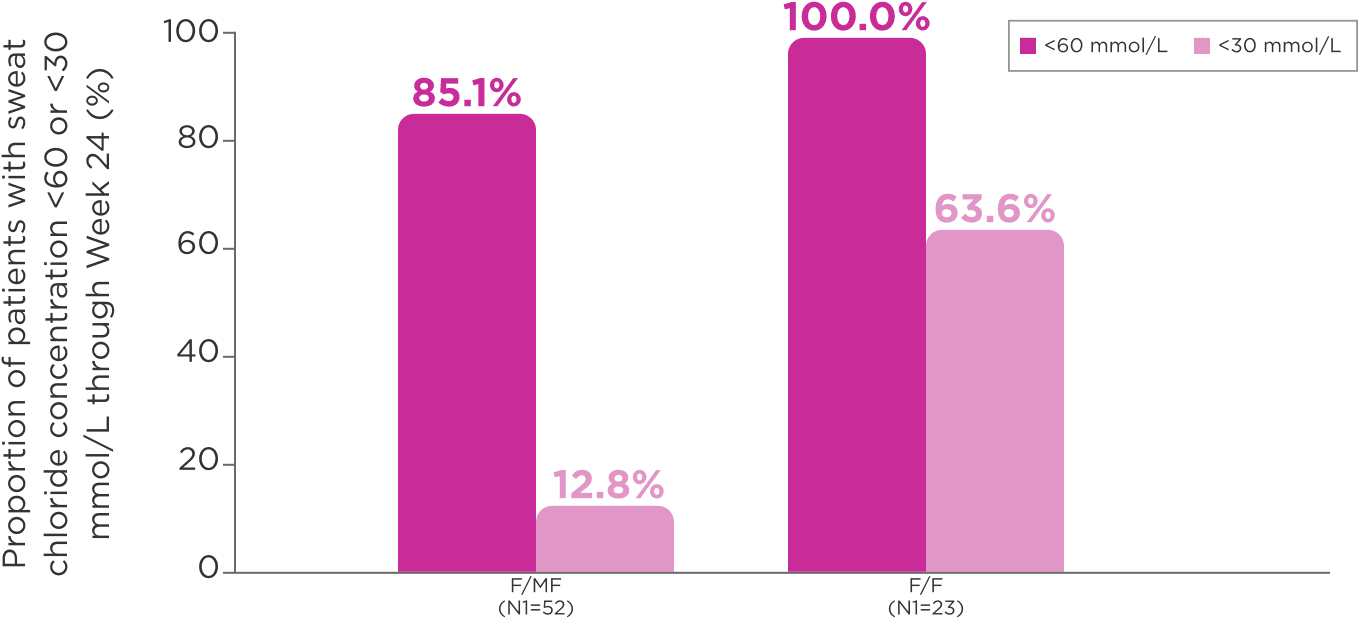
- 1 patient (1.4%) in the study, of F/MF genotype, had a sweat chloride concentration <60 mmol/L at baseline3
fPercentages were calculated by dividing n (the number of patients with sweat chloride concentration below the indicated threshold at week 24) by N1, where N1 is the number of patients with evaluable data. Patients with missing data were considered missing at random and were not counted in the denominator.
DECREASE IN MEAN LCI2.52
LS-mean absolute change from baseline through Week 24 (95% CI: -1.01, -0.66)
Mean (SD) at baseline: 8.41 units (1.48)
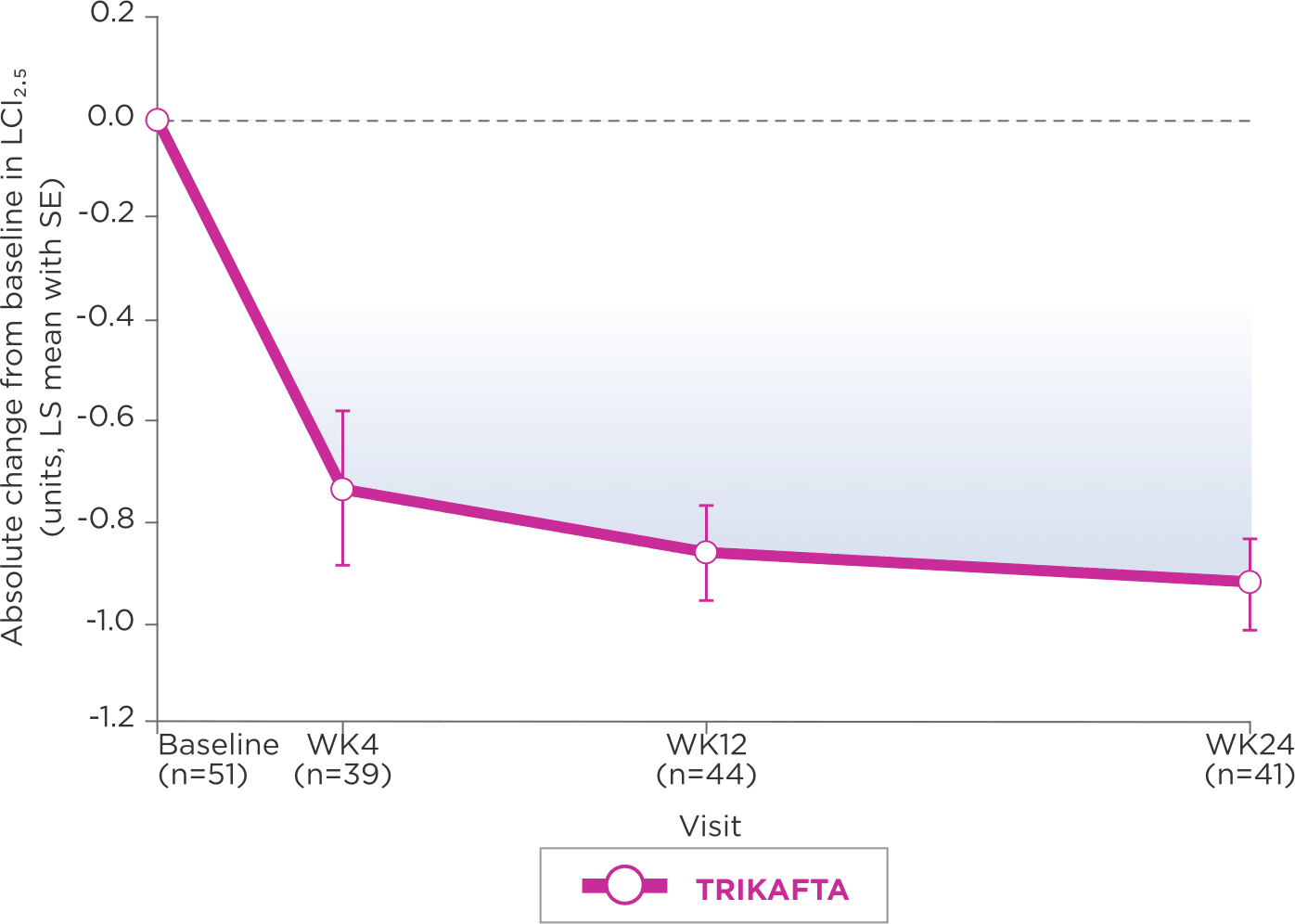
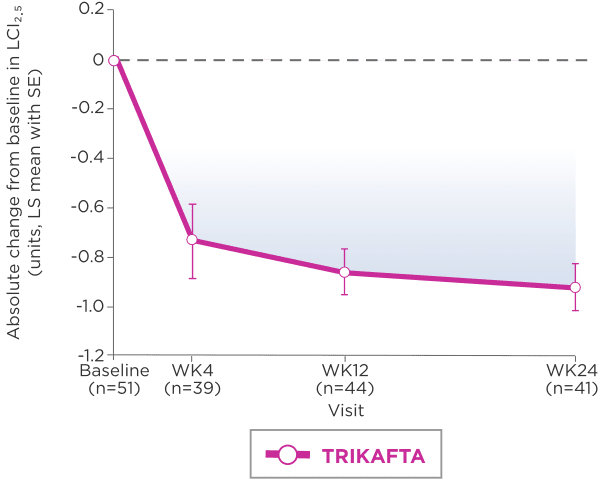
A Subgroup analysis of LCI2.5 by genotype indicates that results for patients with F/F genotype are consistent with those of patients with F/MF genotype3
Lung clearance index (LCI), derived from multiple breath washout tests, is an established research outcome for individuals with CF. It involves following an inert tracer gas washed out from the lungs during relaxed tidal breathing. Key advantages of LCI over ppFEV₁ include an increased sensitivity to early changes in airway obstruction and ability to be performed repeatedly even in very young children with growing lungs.5
gAt screening, LCI2.5 was assessed in patients ages 3 to 5 years only.
hIn Trial 4, mean baseline LCI2.5 was 8.41 units (SD, 1.48) for patients receiving TRIKAFTA.
iLCI represents a measure of the number of times the volume of a gas in the lung at the start of a washout must be turned over to wash out tracer gas to the predefined endpoint.5
Select Additional Trial Outcomes2

CHANGE IN BMI
(95% CI: -0.10, 0.17)
Mean (SD) at baseline: 15.79 kg/m² (1.06)
CHANGE IN BMI-FOR-AGE Z-SCORE
(95% CI: 0.0, 0.20)
Mean (SD) at baseline: 0.09 (0.85)
ALT, alanine aminotransferase; AST, aspartate aminotransferase; CI, confidence interval; F/F, homozygous for the F508del mutation; F/MF, heterozygous for the F508del mutation and a minimal function mutation; GGT, gamma-glutamyl transferase; LS, least squares; PEx, pulmonary exacerbation; SE, standard error; TEAE, treatment-emergent adverse event; ULN, upper limit of normal; WK, week.
IMPORTANT SAFETY INFORMATION
WARNINGS AND PRECAUTIONS
Elevated Transaminases and Hepatic Injury
- Liver failure leading to transplantation has been reported in a patient with cirrhosis and portal hypertension while receiving TRIKAFTA. Avoid use of TRIKAFTA in patients with pre-existing advanced liver disease (e.g., as evidenced by cirrhosis, portal hypertension, ascites, hepatic encephalopathy) unless the benefits are expected to outweigh the risks. If used in these patients, they should be closely monitored after the initiation of treatment
- Isolated elevations of transaminases or bilirubin have been observed in patients with CF treated with TRIKAFTA. In some instances, transaminase elevations have been associated with concomitant elevations in total bilirubin and/or international normalized ratio (INR) and have resulted in patients being hospitalized for intervention, including in patients without a history of pre-existing liver disease
INDICATIONS AND USAGE
TRIKAFTA is indicated for the treatment of cystic fibrosis (CF) in patients aged 2 years and older who have at least one F508del mutation in the cystic fibrosis transmembrane conductance regulator (CFTR) gene or a mutation in the CFTR gene that is responsive based on in vitro data.
If the patient’s genotype is unknown, an FDA-cleared CF mutation test should be used to confirm the presence of at least one F508del mutation or a mutation that is responsive based on in vitro data.
- Assessments of liver function tests (ALT, AST, and bilirubin) are recommended prior to initiating TRIKAFTA, every 3 months during the first year of treatment, and annually thereafter
- In the event of significant elevations in liver function tests, e.g. ALT or AST >5x the upper limit of normal (ULN) or ALT or AST >3x ULN with bilirubin >2x ULN, dosing should be interrupted and laboratory tests closely followed until the abnormalities resolve. Following the resolution of liver function test elevations, consider the benefits and risks of resuming treatment
- For patients with a history of hepatobiliary disease or liver function test elevations, more frequent monitoring should be considered
Hypersensitivity Reactions, Including Anaphylaxis
- Hypersensitivity reactions, including cases of angioedema and anaphylaxis, have been reported in the postmarketing setting. If signs or symptoms of serious hypersensitivity reactions develop during treatment, discontinue TRIKAFTA and institute appropriate therapy. Consider the benefits and risks for the individual patient to determine whether to resume treatment with TRIKAFTA
Concomitant Use With CYP3A Inducers
- Exposure to ivacaftor is significantly decreased and exposure to elexacaftor and tezacaftor are expected to decrease by the concomitant use of strong CYP3A inducers, which may reduce the therapeutic effectiveness of TRIKAFTA. Co‑administration with strong CYP3A inducers is not recommended
Concomitant Use With CYP3A Inhibitors
- Exposure to elexacaftor, tezacaftor, and ivacaftor are increased when co-administered with strong or moderate CYP3A inhibitors. The dose of TRIKAFTA should be reduced when used concomitantly with moderate or strong CYP3A inhibitors
Cataracts
- Cases of non-congenital lens opacities have been reported in pediatric patients treated with ivacaftor-containing regimens. Baseline and follow‑up ophthalmological examinations are recommended in pediatric patients initiating treatment with TRIKAFTA
ADVERSE REACTIONS
Serious Adverse Reactions
- Serious adverse reactions that occurred more frequently in patients treated with TRIKAFTA compared to placebo were rash (1% vs <1%) and influenza (1% vs 0)
Most Common Adverse Reactions
- The most common adverse reactions occurring in ≥5% of patients treated with TRIKAFTA (N=202) and higher than placebo (N=201) by ≥1% in the 24-week placebo-controlled, parallel-group Phase 3 trial (Trial 1) were headache, upper respiratory tract infection, abdominal pain, diarrhea, rash, alanine aminotransferase increased, nasal congestion, blood creatine phosphokinase increased, aspartate aminotransferase increased, rhinorrhea, rhinitis, influenza, sinusitis, and blood bilirubin increased
- The safety profile for the patients with CF receiving TRIKAFTA (N=55) enrolled in the 4-week, randomized, double-blind, active-controlled Phase 3 trial (Trial 2) was similar to that observed in Trial 1
- The safety profile in patients age 6 through 11 years from an open-label trial (Trial 3; N=66) was similar to that observed in Trial 1. The safety profile in patients age 2 through 5 years from an open-label trial (Trial 4; N=75) was similar to that observed in Trial 1
USE IN SPECIFIC POPULATIONS
Pediatric Use
- The safety and effectiveness of TRIKAFTA in patients with CF younger than 2 years of age have not been established
Click here to access full Prescribing Information for TRIKAFTA.
References:
1. TRIKAFTA [prescribing information]. Boston, MA: Vertex Pharmaceuticals Incorporated; August 2023. 2. Goralski JL, Hoppe JE, Mall MA, et al. Phase 3 open-label study of elexacaftor/tezacaftor/ivacaftor in children aged 2 to 5 years with cystic fibrosis and at least one F508del allele. Am J Respir Crit Care Med. Final MS pp1-35, Feb. 27, 2023. 3. Goralski JL, Hoppe J, Mall MA, et al. Phase 3 open-label study of elexacaftor/tezacaftor/ivacaftor in children aged 2 to 5 years with cystic fibrosis and at least one F508del allele. Online data supplement. Am J Respir Crit Care Med. pp1-35, Feb. 27, 2023. 4. A phase 3 study evaluating the safety, tolerability, and pharmacokinetics of elexacaftor/tezacaftor/ivacaftor triple combination therapy in cystic fibrosis subjects 2 through 5 years of age. European Union Clinical Trials Register. Updated October 21, 2021. Accessed March 1, 2024. https://www.clinicaltrialsregister.eu/ctr-search/rest/download/result/zip/pdf/2020-002251-38/1 5. Horsley A. Lung clearance index in the assessment of airways disease. Respir Med. 2009;103(6):793-799. doi:10.1016/j.rmed.2009.01.025